I just arrived in LA for the IPAM Workshop on Algorithmic Challenges in Protecting Privacy for Biomedical Data. I co-organized this workshop with Cynthia Dwork, James Zou, and Sriram Sankararaman and it is (conveniently) before the semester starts and (inconveniently) overlapping with the MIT Mystery Hunt. The workshop has a really diverse set of speakers so to get everyone on the same page and anchor the discussion, we have 5 tutorial speakers and a few sessions or shorter talks. The hope is that these tutorials (which are on the first two days of the workshop) will give people some “common language” to discuss research problems.
The other big change we made to the standard workshop schedule was to put in time for “breakout groups” to have smaller discussions focused on identifying the key fundamental problems that need to be addressed when thinking about privacy and biomedical data. Because of the diversity of viewpoints among participants, it seems a tall order to generate new research collaborations out of attending talks and going to lunch. But if we can, as a group, identify what the mathematical problems are (and maybe even why they are hard), this can help identify the areas of common interest.
I think of these as falling into a few different categories.
- Questions about demarcation. Can we formalize (mathematically) the privacy objective in different types of data sets/computations? Can we use these to categorize different types of problems?
- Metrics. How do we formulate the privacy-utility tradeoffs for different problems? What is the right measure of performance? What (if anything) do we lose in guaranteeing privacy?
- Possibility/impossibility. Algorithms which can guarantee privacy and utility are great, but on the flip side we should try to identify when privacy might be impossible to guarantee. This would have implications for higher-level questions about system architectures and policy.
- Domain-specific questions. In some cases all of the setup is established: we want to compute function F on dataset D under differential privacy and the question is to find algorithms with optimal utility for fixed privacy loss or vice versa. Still, identifying those questions and writing them down would be a great outcome.
In addition to all of this, there is a student poster session, a welcome reception, and lunches. It’s going to be a packed 3 days, and although I will miss the very end of it, I am excited to learn a lot from the participants.
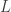 , starting from that position. Just a brief glance at the physical process above shows how simple that model is. For the purposes of statistics, it may be enough, but here are some complications that I can see, from knowing almost no physics and biology:
, starting from that position. Just a brief glance at the physical process above shows how simple that model is. For the purposes of statistics, it may be enough, but here are some complications that I can see, from knowing almost no physics and biology: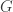 , the length of the genome,
, the length of the genome, 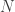 , the number of reads. The questions is how should these depend on each other? David presented an theoretical analysis of the reconstruction question under rather restrictive assumptions (bases are i.i.d., reads are noiseless) and showed that there is a threshold on the number of reads for successful reconstruction with high probability. That is, there is a number
, the number of reads. The questions is how should these depend on each other? David presented an theoretical analysis of the reconstruction question under rather restrictive assumptions (bases are i.i.d., reads are noiseless) and showed that there is a threshold on the number of reads for successful reconstruction with high probability. That is, there is a number 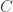 such that
such that  reads can reconstruct the sequence with high probability. The number
reads can reconstruct the sequence with high probability. The number  .
.