I’ll follow up with some blogging about the talks at Allerton a bit later — my notes are somewhat scattershot, so I might give a more cursory view than usual. Overall, I thought the conference was more fun than in previous years: the best Allerton to date. For now though, I’ll blog about the plenary.
David Tse gave the “director’s cut” of his recent work (with Motahari, Bresler, Bresler, and Ramchandran) on a information theoretic model for next-generation sequencing (NGS). In NGS, many copies of a single genome are chopped up into short chunks (say 100 base pairs) of reads. The information theory model is a very simplified abstraction of this process — a read is generated by choosing uniformly a location in the genome and producing the 100 bases following that position. In NGS, the reads overlap, and each nucleotide of the original genome may appear in many reads. The number of reads in which a base appears is called the coverage.
So there are three parameters, 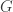
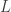
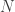
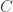


This model is very simple and is clearly not an accurate model for genome sequencing. David began his talk by drawing a grand analogy to the DMC — his proposition is that this approach will be the information theory of genome sequencing. I have to say that this sounds appealing, but it looks at a rather specific problem that arises in NGS, namely assembly. This is a first step towards building one abstract theory for sequencing and while the model may be simple, the results are non-trivial. David also presented some evidence about how real DNA sequences have features (long repeats) which make problems for greedy assemblers but can be handled by more complex assemblers based on deBruijn graphs. They can also handle noise in the form of i.i.d. erasures. What this analysis seems to do is point to features about the data that are problematic for assembly from an information-theoretic standpoint — this is an analysis of the technological process of NGS rather than saying that much about biology.
I’ve been working for the last year (more off than on, alas) on how to understand certain types of NGS data from a statistical viewpoint. I’ll probably write more about that later when I get some actual understanding. But a central lesson I’ve taken from this is that the situation is quite a bit different than it was when Shannon made a theory of communication that abstracted existing communication systems. We don’t have nearly as good an understanding NGS data from an engineering standpoint, and the questions we want to answer from this data are also unclear. Assembly is one thing, but if nothing else, this theoretical analysis shows that the kind of data we have is often insufficient for “real” assembly. This correlated with practice, as many assemblers produce large chunks of DNA, called contigs, rather than the full organism genome. There are many interesting statistical questions to explore in this data — what can we answer from the data without assembling organisms?

Although genome sequencing is certainly a general problem like the problem of communication, I think assembly is specific to shotgun technologies, right? For example, the DNA transistor (http://researcher.watson.ibm.com/researcher/view_project.php?id=1120) would not require assembly.
Yep, that racheting thing is pretty awesome and would eliminate some of these issues. However, it’s a little unclear how well it would work with heterogeneous samples with many organisms in one sample — with shotgun it’s just blend-n-go, which is also part of the appeal, together with lowering costs.